The Data Collection Roadmap
We initially help clients develop their unique Data Collection Roadmap. This is a multiyear plan to design and build interoperable registries and RWD studies for their target rare diseases across Europe. This will provide a unified data collection program to complement and expand on Clinical Trials.
Europe is a collection of large/small and rich/poor counties all with different health care policies. This presents unique challenges to developing an interoperable clinical data platform. A single European registry is not practical. Clinicians will always need to collect their own data to meet local and national research. Most registry owners will require the ability to extend the registry to support their own national data collection needs.
The Roadmap will communicate the long-term path to support the development of an interoperable European Federated Registry for the benefit of all stakeholders. It will define:
- The Model Registry to be used in countries without a sustainable well-established registry.
- The process that industry will use for sharing proprietary data with the registry and the funding of Centres to sponsor studies.
- The structure of the Federated Registry and the Umbrella Patient Organization
- The structure of cooperating agreements between pharma, patient organizations and researchers.
- The features required of any third-party registry to be considered for sponsorship. This will eliminate funding “dead end data” where the minimum dataset is not supported.
- Ownership of the registry data and the process where an independent scientific committee control the scientific direction of the Model and Federated registry.
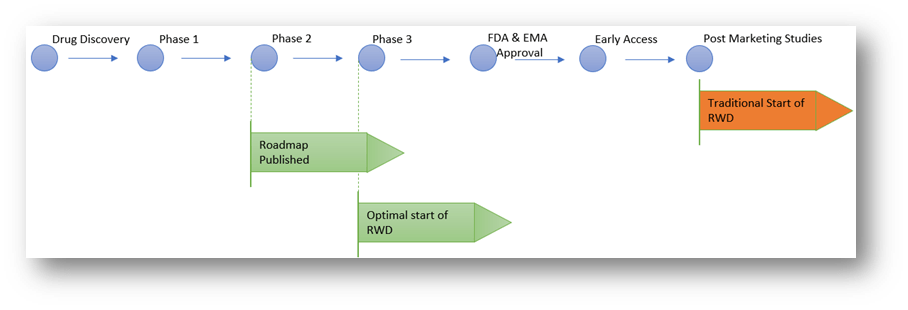
Timeline for roadmap and RWD
We outline above the suggested points within a classical drug development timeline for publishing the Data Collection Roadmap and the RWD platform:
Traditionally RWD does not commence until well after drug approval. We however suggest a more proactive approach:
- Pharma should publish their Data Collection Road Map as soon as possible, ideally during Phase 2. This will provide the research community with an indication of their data collection goals and possible sponsorship opportunities.
- The RWD program should commence at the start of Phase 3 with a Natural History Study. It can be based on the dataset proposed for the Clinical Trial. It should be designed to reflect the natural history of the disease for all patients with the target disease. This will include patients at different stages in the disease development and with different comorbidities.
The protocol for the Natural History Study will be less rigid than the Phase 3 Clinical Trial.
The Clinical Trial would for example specify a minimum number of encounters per year and have tightly specified inclusion and exclusion criteria for subjects. Because of this, the number of patients enrolled in a natural history study should be much larger than in the related clinical trial.
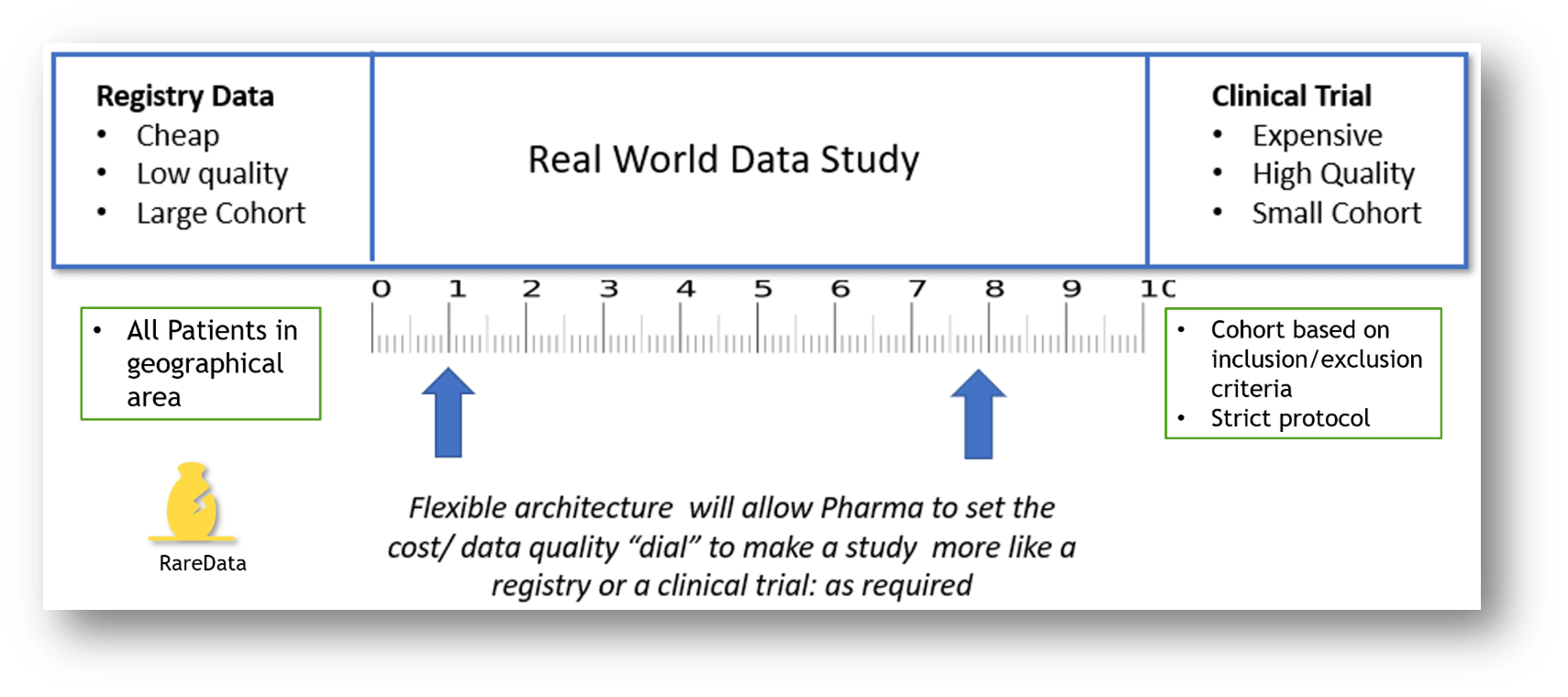
This RWD study will require the cooperation of researchers as outlined in the Data Collection Roadmap. RWD fills the gap between low-cost, low-quality registry data and high-cost high quality clinical trials data
By independently specifying the dataset and the data collection platform, pharma can limit the involvement of an expensive Contract Research Organization (CRO) to just remote monitoring. This can lead to very significant financial savings as the average cost of a Phase 3 orphan drug trial is $30M (source Orphanet Journal of Rare Diseases 2019).